

条件及び期限付制度が適用された再生医療等製品のうち、2023年にハートシート、2024年にコラテジェンが条件及び期限付の期限を迎えました。両製品の動向に注目が集まりましたが、ハートシートは正式承認申請を行うも有効性が認められないとして正式承認には至らず、またコラテジェンは一旦は正式承認申請を行うも、市販後調査で治験の結果を再現できなかったとして申請を取り下げと大方の期待を外れる結果となり、改めて本制度の意義を問う声も上がっています。
そこで、条件及び期限付承認制度のこれまでを振り返りながら、改めて本制度について考えます。
制度導入の経緯
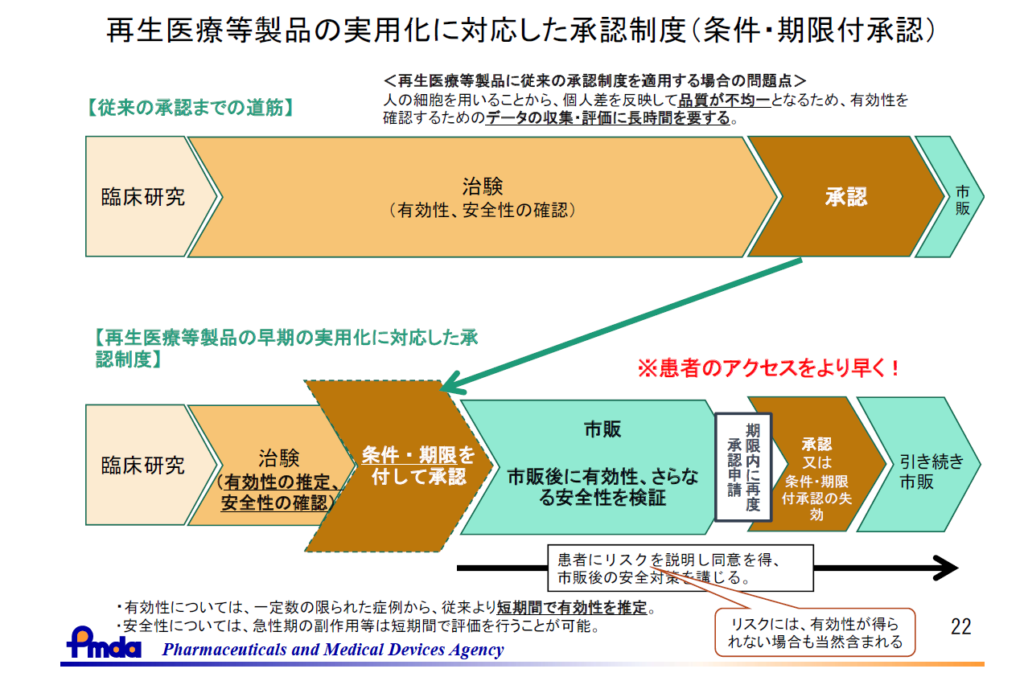
2014年11月に薬事法が「医療機器等の品質、有効性及び安全性の確保等に関する法律」(以下、薬機法) に改正されました。その法律の中で、医薬品や医療機器とは別に「再生医療等製品」が新たに定義されるとともに、その特性を踏まえて「均質でない再生医療等製品については、有効性が推定され、安全性が確認されれば、条件及び期限付きで特別に早期に承認できる仕組み」として、条件及び期限付承認制度が導入されました。従来の承認制度と比較して早い段階で承認されることから、早期承認制度と呼ばれることもあります。

細胞や遺伝子を用いる再生医療等製品では、原材料となる細胞にばらつきがあることから製品は不均一であることが多く、また希少疾患を対象とすることが多いため、有効性の確認に必要な症例数の治験を行うには従来の医薬品に比べて時間を要することが考えられます。
そこで従来の治療法では治療ができない患者さんに、安全性を確保しつつ、かつできるだけ早く届けることを可能とするために、販売先を専門的な医師や設備を有する医療機関等に限定する条件と、原則7年以内の期限付きで、「仮免許」として承認を与える制度として導入されました。
条件及び期限付承認として承認された製品は、期限内に有効性・安全性についての資料をもって再度申請し「正式承認」を取得する必要があり、取得できない場合は承認取り消しとなります。
条件及び期限付として承認を取得した製品
2024年7月現在、条件及び期限付承認制度の適用として承認を取得した再生医療等製品は、ハートシート(テルモ)、ステミラック注(ニプロ)、コラテジェン筋注用(アンジェス)、デリタクト注(第一三共)、アクーゴ脳内移植注(サンバイオ)の5製品です(ただし2024年7月現在、アクーゴは製品出荷については未許可)。それぞれ条件として①専門の医療機関、医師、および管理体制のもとで使用すること、②条件及び期限付承認の期間中は症例全例を対象として評価を行うことが付されており、期限として5~7年が設定されています。
ハートシートは、承認を受けた当初は期限を5年間とされていましたが、その後医療機関との契約に時間を要したこと等により十分な症例が集まっていないことから期限が3年間延長されています。
2023年9月に期限を迎えたハートシートは同年9月に正式承認申請を行いました。しかしながら、2024年7月19日の薬事審議会において、主要評価項目である心臓疾患関連死までの期間、および副次評価項目である心機能に関するLVEFの改善などについて対照群に対して優越性は示されないとして、正式承認には至りませんでした。
一方でコラテジェンは、2023年5月に「従前申請の治験結果の再現性が確認できたと判断」として正式承認申請を行いましたが、その後2024年6月に「非盲検下で実施した市販後調査では、二重盲検の国内第Ⅲ相臨床審成績を再現できなかった」として、条件及び期限付承認および正式承認申請を取り下げ、コラテジェンの販売を終了することを発表しました。
ステミラックは2025年12月、デリタクトは2028年6月にそれぞれ期限を迎えます。
| 製品名/製造販売業者 | 対象疾患/製品 | 承認日 | 期限 |
| ハートシート® /テルモ | 虚血性心疾患による重症心不全 /自家骨格筋由来細胞シート | 2015年9月18日 | 5年間 ※その後3年間の延長 |
| ステミラック®注 /ニプロ | 脊髄損傷に伴う神経症候及び機能障害 /自家骨髄由来間葉系幹細胞 | 2018年12月28日 | 7年間 |
| コラテジェン®筋注用 /アンジェス | 慢性動脈閉塞症 /HGFプラスミドベクター | 2019年3月26日 | 5年間 |
| デリタクト®注 /第一三共 | 悪性神経膠腫 /腫瘍溶解性ウイルス | 2021年6月11日 | 7年間 |
| アクーゴ®脳内移植注 /サンバイオ | 外傷性脳損傷に伴う慢性期の運動麻痺 /他家骨髄由来加工間葉系幹細胞 | 2024年6月19日 | 7年間 |
条件及び期限付承認制度の問題点
条件及び期限付承認制度は日本が世界に先駆けて導入した制度であり、制定当時は海外からも大変注目されました。一方で、国内企業からは審査に対する要望や、また海外を中心に制度そのものに対する批判的な意見も寄せられました。
①審査に対する要望
条件及び期限付承認制度は、申請者が条件及び期限付承認として申請するわけではなく、申請後の審査の中で、規制当局が申請データの内容により通常承認とするか条件及び期限付承認とするかを判断します。
通常承認と条件及び期限付承認では販売開始後の対応が大きく異なるため、製薬企業側からはどのような場合に条件及び期限付承認が適用されるかの「予見性の確保」を求める声が上がっていました。また、条件及び期限付承認の要件のひとつである「有効性が推定されること」についても、何をもって推定されるとするかが曖昧であり、明確にしてほしいとの意見もあります。
これらの意見に対し、2024年3月に厚労省より「再生医療等製品に係る条件及び期限付承認並びにその後の有効性評価計画策定に関するガイダンス」が発出され、その中で条件及び期限付承認の適用対象、開発における留意事項、製造販売後臨床試験のデザインなどに対する基本的な考え方が示されています。また有効性の評価については、同じく2024年3月に間葉系幹細胞を対象として「ヒト由来の間葉系幹細胞若しくは間葉系間質細胞を原料とするヒト細胞加工製品の条件及び期限付承認並びにその後の有効性評価計画に関する評価指標」が発出され、有効性を示すための評価計画や製品の不均一性に対する考え方などが示されています。
②制度に対する批判
本制度に対しては海外を中心に批判的な意見も寄せられ、世界的な学術論文誌であるNature誌やNature Medicine誌,およびScience誌がそれぞれ本制度についての懸念点を指摘する記事を掲載しています。
Nature Medicine誌では、本制度の施行前の2013年に本制度について取り上げ、「患者にとっては費用を負担して有効性が証明されていない治療の被験者になることになる」「治験のようにランダム化や盲検化ができず、また患者がプラセボ効果を得る可能性も高い」との懸念を述べています(Nature Medicine 19, 510(2013))。
条件及び期限付承認の第1号であるハートシートの承認後の2015年12月には、Nature誌上で「日本は臨床試験の費用を患者に負担させるという実証のない制度を導入した」を副題として、通常は開発企業が負担する治験の費用を患者および国民保険が負担している点に加え、再審査時の評価が甘ければ効果のない医薬品であふれてしまうため正式承認申請時の審議を厳格に行うべきであることを指摘しています(Nature 528, 163-164(2015))。
さらにNature誌はステミラックが条件及び期限付承認を取得した際にも、” Japan should put the brakes on stem-cell sales (日本は幹細胞の販売にブレーキをかけるべきだ)”というタイトルで、症例数が少なく、かつ比較対照群が置かれていない臨床試験では適切な評価ができないこと、またステミラックの有効性についての科学的根拠の乏しさについて指摘する記事を掲載しています(Nature 565, 535-536(2019))。
またScience誌は本制度を国際的な経済競争に勝つための政府による規制の緩和として、安全性と有効性を確保するための基準を定めるのが政府の役割であり、ある国において経済的、政治的理由により規制を緩和した場合、その国だけにとどまらず連鎖的に世界レベルで再生医療の分野に予期せぬ有害な結果をもたらす可能性があるとして警鐘を鳴らしています’(Science 365, 644-646(2019))。
これらの指摘に対して、高橋政代 理化学研究所プロジェクトリーダー(当時)は、新しいシステムはそういった危険性ははらむものの、ハートシート®は標準的な治療では効果がない重い心臓機能障害に対するもので、心臓移植のドナー不足の問題がより顕著な日本では唯一の治療法であり、それをタイムリーに提供することは患者への利益のためであり、製薬企業の利益のためではない旨の見解を日本再生医療学会雑誌中で述べています。
また日本再生医療学会は、「日本の再生医療等製品承認プロセスに関する日本再生医療学会の考え方」の声明を発表し、有効性が合理的に推定できることが重要であり、議論のために必要な可能な限りの情報開示が必要であることについてはNature誌と同意見とする一方で、患者数が少ない疾患では、投与群と非投与群を比較し、統計的に有効性を確認するための治験参加者数を揃えることが難しく、必ずしもすべての製品でRCTが必須とは考えていないこと、学会主導による全国的なデータベースとして『再生医療等製品使用データ登録システム(NRMD/PMS)』の構築・運用を開始しており、データを蓄積することで、透明性の高いデータ評価を行うことができること、また、これまでの治療手段とは異なった特性を持つ再生医療等製品の承認制度には、再生医療等製品の特性を鑑みた新しいアプローチも必要であることを述べています。
開発企業および医療現場からの意見
前述のように本制度は治験費用を患者に負担させているに等しい点が批判される一方で、開発する企業側のリスクを低減させることで新しい再生医療等製品の開発が活性化されるという側面もあり、特に資金力に劣るベンチャー企業の中には開発上の戦略として積極的に本制度の適用を狙う企業もあります。
一方ですでに再生医療等製品5品目の承認を取得しているジャパン・ティッシュエンジニアリング(J-TEC)社は、最初から条件及び期限付承認を狙った開発は本質的ではなく、J-TEC社ではそのような方針は取らないことを再生医療学会などの講演において述べています。
また自家骨格筋由来細胞製品の開発を行うイノバセル社も、条件及び期限付承認制度は非常に良い制度であるとした上で、有効性のエビデンスに基づいた通常承認を受けた製品の方が実際に使用する医師にとっても治療を受ける患者にとっても望ましいとの判断から、通常承認を目指す方針を取ることを述べています。
医療現場側の意見としては、日経バイオテク「再生医療等製品の早期承認制度をどう使うか」の中で”「医療現場は本当に効くのかどうかを評価して使うかどうかを決める。早期承認を取得して大きく売れている製品はこれまでに無い。早ければいいというわけではなく、本当に効くかどうかが重要だ」”との意見が紹介されており、”一定の規模の臨床試験を実施して、明確な有効性のエビデンスを示し、医療現場に使ってもらうというのが、結局は一番の近道なのだという認識が広がりつつある”とされています。
条件及び期限付承認制度のこれから
条件及び期限付承認制度は、新しい治療をいち早く患者さんのもとへ届けるために作られた制度です。一方でこれまで述べてきたように本制度が本当に患者さんの利益になっているのか、開発企業側の利益優先となっていないかとの疑問視する声があるのも事実です。今回ハートシート、コラテジェンが立て続けに正式承認を取得できなかったことで、それらの声がより大きくなるかもしれません。
本制度が今後も必要かについては賛否両論あると思いますが、患者さん側の立場に立って考えると、個人的意見としては条件及び期限付承認制度は今後も必要な制度であり、開発企業および審査当局側の取組みを踏まえると将来的には患者さんの利益にとってより有用な制度となると考えています。
ハートシート、コラテジェンの対象疾患がそれぞれ「薬物治療や侵襲的治療を含む標準治療で効果不十分な虚血性心疾患」、「標準的な薬物治療の効果が不十分で血行再建術の施行が困難な慢性動脈閉塞症」であるように、再生医療は既存の治療法では治すことのできなかった疾患やケガを治すことができる可能性のある医療です。
有効性が確認されていない治療を患者さんに提供することはあってはならないことはその通りですが、一方で患者さん側からは、ほかに有効な治療法がない状況で承認まで待っていられない、少しでも可能性があるのであれば治療を受けたいという声を聞くことが多く、本制度が患者さんの希望となっていることも事実です。そのため制度自体は今後も続けるべきだと考えます。
とは言え開発側がそれに甘えることは許されず、「少しでも可能性がある治療」ではなく「本当に有効な治療」を提供する責任があり、Nature誌から指摘のあったような「効果のない医薬品であふれる」ことは避けなければなりません。
患者さんの利益を考えた場合、治療効果、経済的負担の2面を考える必要があります。
まず経済的負担については、健康保険および高額医療制度の適用下では薬価額面ほどの費用負担はないとは言え、有効性が実証されていない製品に対して多少なりとも費用負担を強いることは患者さんにとっては不利益です。しかし企業側の負担を軽減することで開発が促進され新たな治療法が開発されれば、長い目で見れば患者さんの利益につながるとの見方もあります。
企業側の負担軽減のみを考えるのであれば、条件及び期限付承認制度の形ではなく、例えば国立研究開発法人日本医療研究開発機構(AMED)事業として第3相治験の費用を助成するという方法もあるかと思いますが、従来の低分子医薬などと比較して製品が不均一であり(原材料となる細胞によるばらつき)、また作用機序が複雑で正確に把握することが困難な再生医療等製品の性質を踏まえると、被験者の条件などが完全に計画・管理される治験より、一旦承認をして市販後のリアルワールドデータ(病院で日常的に使われることで得られるデータ)として広くデータを収集・評価する方が有用なのかもしれません。
有効性が実証されていない製品であることから、例えば条件付き期限付承認の期間は薬価を低く設定し(例えば製造原価程度)、正式承認時に改めて通常の医薬品と同じ基準で薬価を設定するなどの方法も考えられると思います。
治療効果の面においては、そもそもの本制度のコンセプトを考えると条件付き期限付承認の時点では有効性の推定とならざることは仕方ありません。ただしその推定が実証に確実につながるように、今後も継続して「推定の確度」を高めていく必要があります。
再生医療はまだ歴史が浅い上に製品も多様であり、開発側にも有効性の根拠を適切に示すための解析技術が十分になく、審査側にも評価指標や審査のための知識・経験が足り定ない状況でした。ただし新たな解析技術の開発が日々行われており、また審査側では様々な製品に対応した「次世代医療機器評価指標」の作成が進められており、今後、推定の確度は着実に高まっていくものと考えられます。
「効果のない医薬品であふれる」ことを避けるためには、正式承認の審査を厳格に行うことが必須となります。仮免許とは言え一旦承認したものに対して審査が甘くなることが懸念されていましたが、今回ハートシート、コラテジェンが承認されなかったことは残念ではあるものの、適正な審査が行われたという観点からは評価すべきであると思います。
再生医療等製品の条件及び期限付承認は日本独自の新しい制度であり、前例がないだけにまだ多くの問題点があり、疑問、批判があるのも事実です。しかし開発側・審査側双方の努力により、将来的には患者さんにとって必ず利益になる制度となるものと考えます。