

| 売名 | コラテジェン®筋注用 |
| 一般的名称 | ベペルミノゲン ペルプラスミド (Beperminogene Perplasmid) |
| 製造販売者 販売元 | アンジェス株式会社 田辺三菱製薬株式会社 |
| 対象疾患 | 標準的な薬物治療の効果が不十分で血行再建術の 施行が困難な慢性動脈閉塞症(閉塞性動脈硬化症 及びバージャー病)における潰瘍 |
| 承認日 /保険収載日 | 2019年3月26日(条件及び期限付承認) /2019年9月4日 |
| 保険償還価格 | 60万360円 (保険償還価格の算定) |
| 関連文書 | 添付文書 (PMDAウェブサイト) 審査報告書 (PMDAウェブサイト) 申請資料概要 (PMDAウェブサイト) インタビューフォーム |
【製品概要】
田辺三菱製薬株式会社 コラテジェン情報ページ
コラテジェン®筋注用は(以下、コラテジェン)は、標準的な薬物治療の効果が不十分で血行再建術の施行が困難な慢性動脈閉塞症に対する遺伝子治療用製品であり、日本初の遺伝子治療薬です。血管内皮細胞の増殖作用を有するヒト肝細胞増殖因子(Hepatocyte Growth Factor; HGF))をコードするプラスミドDNAベクターであり、虚血付近の筋肉内に投与することで筋肉細胞内でHGFが産生され、その結果血管新生が誘導されることで虚血肢の潰瘍の症状を改善します。保険償還価格の算定における市場規模予測では、ピーク時の患者数992人、販売金額12億円と予測されています。
製品名(販売名)の由来は、『血管新生により側副血行路 “collateral vessels” を形成することで、虚血状態の改善を図る遺伝子治療用製品 “gene therapy product”』です。
製造販売社であるアンジェス株式会社は、森下竜一 現大阪大学教授が開発を進めていたHGF遺伝子治療薬の実用化を目的に設立された企業です。
2023年5月に正式承認申請を行いましたが、その後治験の成績を再現できなかったとして条件及び期限付承認および正式承認申請を取り下げ、販売を終了することが発表されました。
【対象疾患と作用メカニズム】
対象疾患
標準的な薬物治療の効果が不十分で血行再建術の施行が困難な慢性動脈閉塞症(閉塞性動脈硬化症及びバージャー病)が対象です。閉塞性動脈硬化症は、手足の血管が動脈硬化により血管内にプラークや血栓が生じることで血管が詰まり、血行が妨げられる(=虚血)疾患です。バージャー病は閉塞性血栓血管炎とも呼ばれ、原因は明らかとなっていませんが手足の末梢血管が閉塞し、虚血が起こる疾患です。これらの慢性動脈閉塞症では、手足に十分に血液が供給されないことで、間欠性跛行(足のしびれや痛みにより歩行に障害をきたした状態)や安静時でも痛みを感じるようになり、重症化すると主に足先に潰瘍を生じ、潰瘍が改善されない場合には組織深部の壊死へと進行し、最悪の場合切断が必要となります。閉塞性動脈硬化症では薬物による治療や、血管バイパス手術やカテーテル治療といった血行再建術が行われますが、薬物治療の効果が不十分で、かつ血行再建術が困難な患者が対象となります。
構造および作用メカニズム
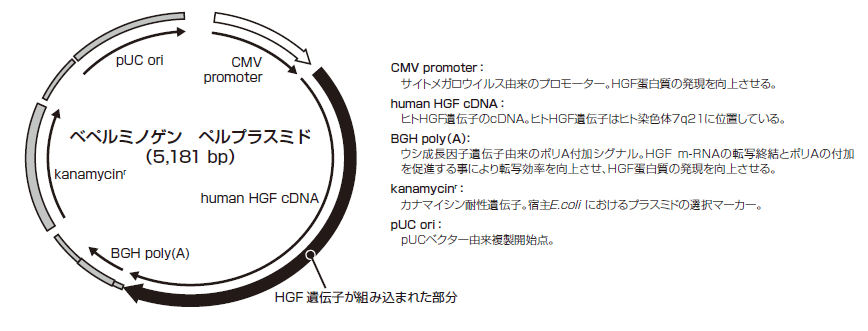
コラテジェン(ベペルミノゲン ベルプラスミド)は、サイトメガロウイルスプロモーター/エンハンサーの制御下でヒトHGFを発現する5,181塩基対からなるプラスミドDNAベクターです。
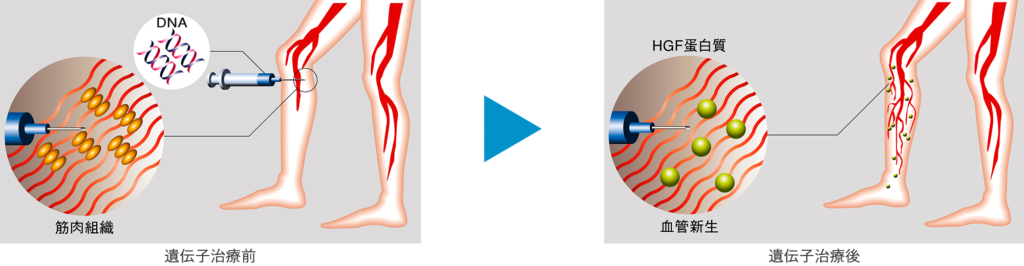
虚血部位に1ヵ所あたり0.5mgずつ8ヵ所(計4mg)を、4週間間隔で2回筋肉内投与します。投与されたコラテジェンは筋肉細胞内に取り込まれた後、転写・翻訳され、HGFタンパクを産生・放出します。放出されたHGFの血管新生作用により、虚血部位の血流が改善され、潰瘍が改善されると考えられています。
他の血管新生因子との比較
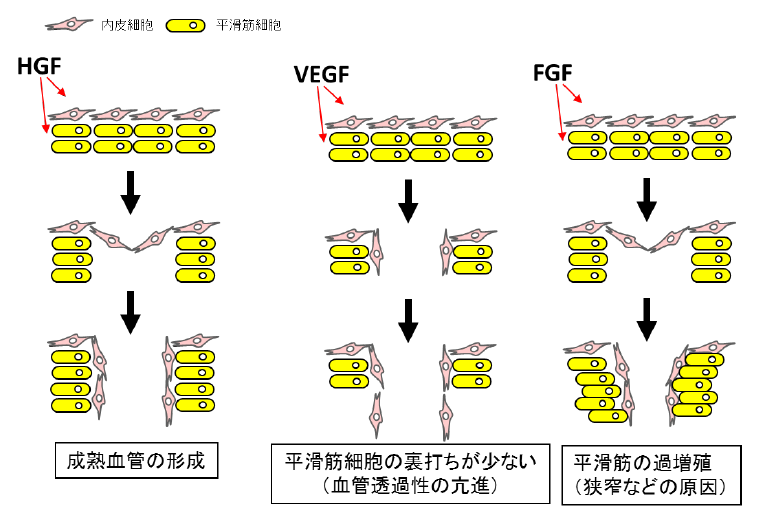
HGF以外の血管新生因子として、血管内皮増殖因子(Vascular Endothelial Growth Factor, VEGF)や線維芽細胞増殖因子(Fibroblast Growth Factor, FGF)が知られています。
血管は内皮細胞と結合組織からなる内膜、平滑筋と弾性線維からなる中膜、疎性結合組織からなる外膜の3層から構成されています。VEGFは主に血管内皮細胞の増殖を促進するため、平滑筋細胞の裏打ちの少ない血管透過性の高い血管(漏れやすい血管)が新生されてしまいます。一方でFGFは血管平滑筋細胞の増殖を促進することから、中膜が厚くなり内腔が狭く詰まりやすい血管となります。これらに対して、HGFは血管内皮細胞の増殖とともに血管平滑筋細胞の遊走を促進することで、機能的な血管が誘導されることが示唆されています。
【条件及び期限付承認】
コラテジェンは、条件及び期限付承認として承認を受けています。条件及び期限は以下の通りとなっています。
条件:
1. 重症化した慢性動脈閉塞症に関する十分な知識・治療経験を持つ医師のもとで、創傷管理を複数診療科で連携して実施している施設で本品を使用すること。
2. 条件及び期限付承認後に改めて行う本品の製造販売承認申請までの期間中は、本品を使用する症例全例を対象として製造販売後承認条件評価を行うこと。
期限:
5年間
正式承認審査結果:
2023年5月に「従前申請の治験結果の再現性が確認できたと判断」として正式承認申請を行いましたが、その後2024年6月に「非盲検下で実施した市販後調査では、二重盲検の国内第Ⅲ相臨床審成績を再現できなかった」として、条件及び期限付承認および正式承認申請を取り下げ、販売を終了することが発表されました。
【適応拡大開発状況】
2019年10月より、慢性動脈閉塞症の安静時疼痛を有する患者を対象にした第III相臨床試験が国内で開始されています。試験期間は約2年間、集積患者数は約40例が予定されています。
②米国における下肢潰瘍を有する閉塞性動脈硬化症 後期第Ⅱ相臨床試験
2020年2月より、下肢潰瘍を有する閉塞性動脈硬化症の患者を対象にした第IIb相臨床試験の患者登録が米国で開始されています。観察期間は12ヵ月、目標症例数は60例となっています。