

| 販売名 | ステミラック®注 |
| 一般的名称 | ヒト(自己)骨髄由来間葉系幹細胞 |
| 製造販売者 | ニプロ株式会社 |
| 対象疾患 | 脊髄損傷に伴う神経症候及び機能障害 ただし、外傷性脊髄損傷で、ASIA機能障害尺度がA、B又はCの患者に限る |
| 承認日 /保険収載日 | 2018年12月28日(条件及び期限付承認) /2019年2月26日 |
| 保険償還価格 | 1495万7755円 |
| 関連文書 | 添付文書 (PMDAウェブサイト) 審査報告書 (PMDAウェブサイト) 申請資料概要 (PMDAウェブサイト) 最適使用推進ガイドライン (厚生労働省ウェブサイト) |
【製品概要】
ニプロ社再生医療等製品サイト
ステミラック®注は、脊髄損傷に伴う神経症候及び機能障害の治療を目的とした、自家骨髄由来間葉系細胞製品です。札幌医科大学 神経再生医療学部門の本望修 教授らとニプロ株式会社が共同で開発し、2018年に条件及び期限付承認として、ニプロ社が再生医療等製品の製造販売承認を取得しました。
脊髄損傷に対する世界初の再生医療製品として注目される一方で、治験内容や作用機序の不明瞭さ等について、ネイチャー誌より批判を受けています。
2019年度(販売開始は2019年5月のため、約10ヵ月間)の売上は2.4億円となっています(ニプロ株式会社決算概要)。保険償還価格から算出した推定適用患者数は16人となります。
【対象疾患と作用メカニズム】
対象疾患
脊髄損傷に伴う神経症候及び機能障害が対象です。脊髄は背骨(脊椎)の中にある神経の束で、脳と手足といった各部位の間の信号の伝達を司る重症な組織です。事故やスポーツ中のケガなどにより強い衝撃が脊椎に加わることで脊髄が損傷を受けた状態が脊髄損傷であり、損傷部位や度合いによって手足などにまひや感覚障害が起こります。このような外傷性脊髄損傷の中で、ASIA(アメリカ脊髄障害協会)が定める機能障害尺度のA, B, またはCがステミラック®注の適用対象となります。

また適格基準として、受傷後31日以内を目安に骨髄の採取が可能であること、それに伴い、受傷後2週間以内を目安に札幌医科大学附属病院に転院が可能なこと、骨髄採取に耐えられる全身状態であること等が求められています。
作用メカニズム
ステミラック®注の作用機序としては、「損傷部位へ集積し、神経栄養因子等を介した神経保護作用を引き起こし、免疫調節、神経系細胞への分化、その他複数の機序により神経症候を改善すると推察」と添付文書に記載されています。
また、ラットを用いた動物試験では、血液脊髄関門の修復が移植早期における運動機能の回復に寄与しているとし、さらに微小血管系の修復、損傷軸索の再生、脱髄軸索の再髄化などの協奏的な作用がまひの回復に貢献していることが示唆されています(科学研究費助成事業 研究成果報告書/課題番号26462213)。
【治療の流れ】
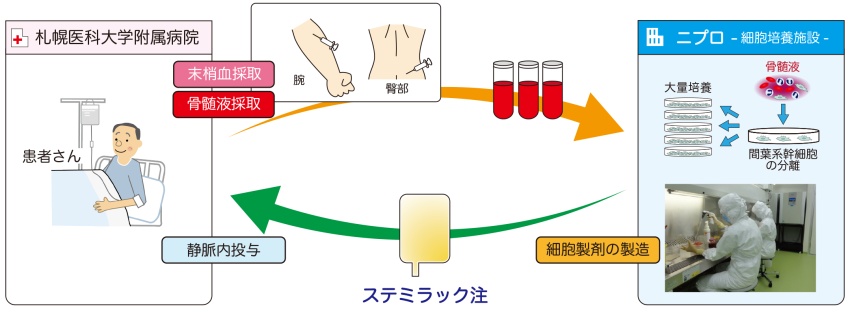
ステミラック®注を用いた治療は、当面は札幌医科大学附属病院のみで行われる見通しとなっています。そのため、治療を受ける患者は、受傷後2週間以内に札幌医科大学附属病院に転院する必要があります。そこでの検査により適格となった場合、受傷後31日以内に骨髄が採取され、その後ニプロ社の培養施設(札幌CPF)において間葉系細胞を分離し、2~3週間かけておよそ1万倍(1億個)にまで培養します。品質試験を経て製品化された後、札幌医科大学附属病院にて静脈注射で投与されます。
ニプロ社は、2020年6月に東京CPFを建設しています。稼働予定は2022年以降ですが、現在の年間最大100患者分の製造から、2024年度には年間最大2500患者分の製造へと、生産体制が大きく増強される計画となっています(ニプロ株式会社 中期経営計画 2020年4月~2025年3月)。
一方で、現在の製造方法では製造原価として3,000万円かかっており、利益の出ない構造となっていることから、原価の低減が大きな課題となっています。
【条件及び期限付承認】
ステミラック®注は、条件及び期限付承認として承認を受けています。条件及び期限は以下のようになっています。
条件:
1. 緊急時に十分対応できる医療施設において、脊髄損傷の診断治療に対して十分な知識・経験を持つ医師のもとで、本品の使用が適切と判断される患者に対して、バイタルサインの確認、臨床検査によるモニタリングや管理等の適切な対応がなされる体制下で本品を使用すること。
2. 条件及び期限付承認後に改めて行う本品の製造販売承認申請までの期間中は、本品を使用する症例全例を対象として製造販売後承認条件評価を行うこと。
期限:
7年間
この期間中に、ステミラック®注の投与を受けない対照群と比較し、有効性を示す必要があります。ニプロ社は予想登録症例数として、適用群:198例、対照群:414例を設定しています(ステミラック®注 審議結果報告書)。
【Nature誌からの批判】
ステミラック®注の承認について、治験の内容の不十分さや有効性の科学的根拠の乏しさなどNature誌が批判的な2つの記事(記事1)(記事2)を掲載しています。ハートシートに続くNature誌からの批判記事ですが、Science誌も日本の早期承認制度に言及しています。
Nature誌の指摘は以下の2点です。
1. 治験の内容の不十分さ
ステミラック®注は、13例の臨床データをもって申請をされ、承認されました。また、通常の医薬品の治験では治療を行わない比較対照群を設定し、さらに心理的なバイアスを取り除くために二重盲検法による無作為化比較臨床試験(RCT; Randomized Controlled Trial)が行われますが、ステミラック®注の治験では、比較対照群が設定されていません。このように症例数が少なく、かつ比較対照群がない試験では適切な評価ができないことをNature誌は指摘しています。
2. 有効性の科学的根拠の乏しさ
ステミラック®注の治験では、投与した13名の患者中12名でASIA機能障害尺度の一段階の改善が見られたとして、その作用機序として「損傷部位へ集積し、神経栄養因子等を介した神経保護作用を引き起こし、免疫調節、神経系細胞への分化、その他複数の機序により神経症候を改善すると推察」としています。しかしながら、たしかに2000年代の前半の研究では、MSCがニューロンのタンパクを発現するなどニューロンの性質の一部を獲得しうることが示されましたが、MSCが機能的なニューロンとして働くことができるという考えは今では多くの研究者によって否定されていること、また、静脈注射により投与されたMSCの大半は肺でトラップされるため、脊髄の損傷部位に達して神経になったとは考えにくいとNature誌は記事の中で述べています。
さらに、これらの臨床データは「宣伝資料として使われることを防ぐため」という理由で公表されておらず、その点もNature誌は批判しています。
また、日本国内においても、民間の医薬品監視団体である「薬害オンブズパースン会議」より、ステミラック注に関する承認を取り消し、臨床試験に戻し有効性・安全性を再確認することを求める要望書が厚生労働省に提出されています。
これらに対し日本再生医療学会は、「日本の再生医療等製品承認プロセスに関する日本再生医療学会の考え方」の声明を発表し、有効性が合理的に推定できることが重要であり、議論のために必要な可能な限りの情報開示が必要であることについてはNature誌と同意見とする一方で、患者数が少ない疾患では、投与群と非投与群を比較し、統計的に有効性を確認するための治験参加者数を揃えることが難しく、必ずしもすべての製品でRCTが必須とは考えていないこと、学会主導による全国的なデータベースとして『再生医療等製品使用データ登録システム(NRMD/PMS)』の構築・運用を開始しており、データを蓄積することで、透明性の高いデータ評価を行うことができること、また、これまでの治療手段とは異なった特性を持つ再生医療等製品の承認制度には、再生医療等製品の特性を鑑みた新しいアプローチも必要であることを述べています。